|
Research & Focus
Genomic data has never been more accessible, and this is leading to many new and exciting discoveries in fields traditionally not focused at the molecular level. Importantly, although tool and approach development is typically initialized in highly-funded model systems, with effort this can often be subsequently applied to any species that could benefit from the technology. Examples may include identification of markers associated with important traits, high-throughput genotyping for parentage or genetic stock identification, or surveying the molecular phenotypic responses to environmental stimuli. Throughout my research career, I have been fortunate to have studied in many leading laboratories with great scientists. During this time, I have always been fascinated by the rapid advances in biological or ecological knowledge that can come from applying frontline genetic approaches to non-model species. Although conservation genetics or aquaculture might not receive as much funding as human health or agriculture, the advances that can be made can rapidly progress as they can ‘stand on the shoulders’ of these other fields. One aspect required for such advancement and portability between fields is the use of open data and code. Not only does this improve reproducibility, but it may also facilitate re-analysis of datasets in ways the original generator may have never considered. Said differently, we work best when we work together, and open science can facilitate decentralized collaboration. Therefore, in my work I always put a strong focus on ensuring reproducible analyses, and wherever beneficial, developing more generalized tools that can be reused for other species or purposes. Currently I am working alongside many different research groups as a collaborative partner through my company, Sutherland Bioinformatics. Over the past 10+ years, I have been involved in research in an academic or government setting, and now am applying the skills that I have developed to make the most positive impact possible. General themes of my research that have remained consistent over the years involve population genomics and transcriptomics of salmonids and other fishes, shellfish, invertebrate parasites (namely sea lice), as well as the general application and development of molecular ecology tools. Please get in touch if you’d like to discuss further, or if you think our collaboration could be right for your research program! |

Collecting eDNA samples at Île d'Anticosti as part of the CanadaC3 eDNA project.
Photo: aquapixels https://www.aquapixels.com
Method Development: MapComp
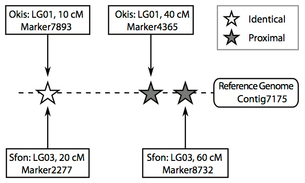
Click image for MapComp paper
Presentation from Coastwide Salmonid Genetics Meeting (2018)
Method Development: GO Trimming

Click image for GO Trimming paper
|
© Ben J G Sutherland 2023 All rights reserved
